
Applying good cell culture practice to novel systems
The principles of good cell culture practice for the growth, banking and use of conventional cell lines is well documented1 2. These guidelines form the foundations for the cell banking procedures at ECACC and underpin the structure of ECACC training courses. The primary goals of good cell culture practice are to prevent cell-line misidentification, contamination with microorganisms, such as mycoplasma and to maintain the genetic and phenotypic stability of cell lines. In good cell culture practice, consideration needs to be given to the developing and sourcing of cell lines, quality control, cryopreservation and transfer of cell lines between laboratories. It is also important that researchers comply with legal and ethical requirements when deriving cell lines from human and animal tissues, use the correct equipment for growth of their cells and be aware of strategies for troubleshooting when problems are encountered.
In the development of new cell lines it is essential that the cells are properly characterised for the expression of expected markers and the cell line is authenticated by testing DNA from the donor through the use molecular techniques to confirm the species. In the case of human cell lines short tandem repeat (STR) DNA fingerprinting should be used to ensure the cell line matches the donor, and does not match any pre-existing cell lines. Attention should be given to regulations such as the human tissue act, ownership and data protection and patient confidentiality, whilst concurrently ensuring that any clinical metadata relevant to the donor is collected and stored safely. Researchers should also strive to give novel cell lines unique names, to avoid confusion with pre-existing cell lines and possible future mix-ups.
Similar levels of attention are required when acquiring cell lines from other laboratories. Any incoming cell lines must be subject to a series tests to confirm species, identity, freedom from microbial contamination including mycoplasmas and until those tests have been completed the cell line must be effectively quarantined.
Care must be made in the appropriate storage and cryopreservation of frozen stocks of cells. It is imperative that the correct rate of freezing is chosen, that vials of cells are stored at a temperature below -130⁰C and ideally Master and Working Cell Banks (MCB and WCB) should be prepared to ensure consistency and reproducibility of cells by providing material of similar passage to application.
Researchers must always be aware of the risks of cross contaminating cultures and there must be strict policies of clearance and segregation in the cell culture lab to ensure that cell lines are not handled in the hood at the same time, cell culture reagents should be aliquoted and dedicated to specific cell lines and never shared between cell lines. Any cell culture laboratory will require a pragmatic regime of regular QC testing and screening of cultures for identity and mycoplasma contamination.
Operator and environmental safety forms a vital core of good cell culture practice. Cellular material from clinical specimens and animal sources may harbour endogenous retroviruses and those derived from cancers may present a risk of tumourogenicity. Cell lines may have been transformed and immortalised by direct intervention and many cell culture regents present toxic and mutagenic hazards. Cell culture should therefore, be performed in appropriate biosafety containment with proper control measures. Typically this means working at containment level 2.
Maintenance of the cellular microenvironment is an important factor in both ensuring cellular proliferation and maintaining cellular phenotype. The use of the correct cell culture medium formulation as described by the supplier or in the literature, and selection of appropriate incubation temperatures and CO2 concentrations is essential. Regular (daily) inspection of cultures by microscopic examination and capture of representative images of the cells is an indispensable procedure as it will give operators early indications of problems with cell growth and potential contamination and cross contamination events.
Current guidelines focus on the growth of continuous, or “immortal” cell lines which offer the main advantage of being relatively easy to grow and robust (by this we mean they can be plated at low densities).
When using these cell lines as models of normal tissue their use is limited by the fact that they are derived from cancers and generally have abnormal karyotypes making them genetically unstable, they tend to exhibit Warburg and Crabtree3 effects, meaning that that the cells tend to ferment rather than obtaining energy from oxidative phosphorylation, using mitochondria as normal tissues do, the cells display increased oxygen dependent glycolysis followed by lactic acid fermentation. This could be a combined effect of the cancerous nature of the cells and the way in which they are grown: at high oxygen concentrations in media and unnaturally high glucose content.
Although it is well known that continuous cell lines lose their differentiated characteristics, their unlimited subculture potential and ready availability means they are suitable for a variety of high throughput applications and are frequently used as cellular models and conventional two dimensional (2D) culture techniques are useful for the expansion of cell stocks and for many simple cell culture experiments.
Induced pluripotent stem cells (iPSC) offer major advantages over conventional cancer derived cell lines and have been available for over 10 years. First developed by Shinya Yamanaka’s lab in Japan4, these cells are derived from adult somatic cells which, under the influence of stem cell transcription factors and chemical agents can be reprogrammed to a stem cell like state, and, in theory, like embryonic stem cells, be differentiated into any tissue. IPSC have huge potential in personalised medicine, diagnostics, research and regenerative medicine as they offer the potential for the development of tissue specific laboratory models from individual disease states and patients. They do not have the same ethical constraints as embryonic stem cells and because, in theory, they can be generated from anyone, their potential supply is limitless.
The disadvantages with iPSC are that, like embryonic stem cells they require a high level of specific technological expertise. Due to their innate “desire” to differentiate quality control of stocks to demonstrate they still express stem cell markers is essential. iPSC are still considered a novel technology and they should be handled with certain caveats. Although theoretically they are pluripotent, it now seems that certain iPSCs have predispositions to differentiate into certain tissues. The wealth of literature on neuronal and cardiomyocyte differentiation from iPSC is large because of the needs of large pharma for toxicity testing.
In the general maintenance of iPSC cultures it is very important to prevent the cells from differentiating. This in itself is challenging because without intervention the cells will do this as a matter of course. To prevent non-specific differentiation the correct medium formulation, must be used and regular sub-culturing and daily medium exchanging is required. There has to be regular confirmation of the expression of pluripotency markers by immunostaining (Immunofluorescence or flow cytometry) or by gene expression (e.g. QRT PCR).
Obtaining vial to vial consistency in iPSC cell banks is difficult to achieve as during culture it is not advisable to generate single cell suspensions and cryopreservation is carried out with colonies, rather than cell suspensions.
There is a genuine concern that the explosion in interest in iPSC cell lines might lead to a new generation of misidentified and cross contaminated cultures. IPSC generation is lengthy and complex and in that process it is essential to keep cultures segregated to prevent potential of cross contamination. It is strongly advised any researchers who are developing iPSCs, particularly in large studies should collect STR profiles of donors and their derived iPSC cell lines and then compare the two to ensure there have been no mix ups or cross contamination events. One way to avoid this risk is to source pre-generated iPSC cell lines from validated collections such as the Sanger HipSci banks that are available from ECACC.
Whether starting with conventional cell lines or iPSC, often the desired end points are differentiated, tissue relevant cultures.
There are many factors that drive cellular differentiation. Firstly there are cell to cell interactions which can be homotypic (between cells of one type, and could be contact mediated by direct cell contact through cell junctions) or they may be heterotypic (via soluble factors from cells of another type, for example paracrine factors from mesenchymal supporting cells). These factors can be reproduced by developing a co-culture systems with mesenchymal fibroblast cells, in for example a Transwell™ system.
Oxygen concentration may be critical to driving differentiation. This can modulate this by using advanced hypoxic cell culture systems, or by increasing or decreasing the depth of medium over the cultures. The shape and polarity of the cells is another important factor. In in vitro systems the shape and polarity can be modulated and altered by the use of scaffolds and hydrogels. Medium formulation and the physical nature and chemical composition of the extracellular matrix will also drive cellular differentiation. In epithelial systems researchers often aim to induce the establishment of basement membrane. This can be reproduced by the use of Matrigel™ or by use of a physical scaffold and cellular environment that encourages the cells to secrete their own basement membrane. Finally, last but not least, the factor of time: many differentiated cultures require several weeks or more to become fully mature.
Three dimensional (3D) Culture systems offer the potential to allow the recapitulation of in vivo - like tissue organisation. Conventional 2D culture on treated plasticware can lead to cells of an undifferentiated phenotype that are no longer representative of the tissue to be studied. The use of scaffolds and hydrogels allow cells to attain the correct orientation and shape and can result in accurate tissue modelling.
It is important that researchers fully validate their putative differentiated cell cultures by looking for surface marker expression confirmation of key functionality. It also is important to show that other markers are absent such as the absence of pluripotency markers and the absence of markers of other differentiated cell types.
The generation of differentiated cultures from iPSC also poses risks from the perspective of cell line validity. A typical iPSC differentiation protocol5 to derive lung epithelial cells from iPSC might at Day 0 with an iPSC Culture. The first step would be step to derive definitive endoderm under the influence of Activin A, this which might take 6 days or more. Then, with the addition of Noggin and the TGFB inhibitor SB431 542 the cells would be driven to anterior foregut for a further 2 or more days.
Cytokines and WNT3a would then be added to drive cells in to a more differentiated state which could take several weeks. Finally is a period of maturation is required before the final phenotype is achieved and at each step it is important to characterise the cell types, looking at the expression of key markers. However, in terms of good cell culture practice this lengthy process could be at risk without proper control. External factors could impact on the outputs of the process. These include prolonged exposure to potentially mycoplasma infected materials, the use of other cell lines in the laboratory and cell culture hoods and environmental contaminants.
There are challenges with the application of good cell culture practices to 3D cell culture systems. Long term 3D co culture models may also be at risk of cross contamination. It is relatively easy to individually identity test component cell lines before the assembly of the complex model, however, at the end of the experiments it is not yet known how easy it will be to identity test co-cultures.
Another challenge associated with complex 3D culture systems is in the imaging of the cultures. In 2D it is relatively easy to get good quality images of the cultures on a day to day basis using a standard inverted phase contrast microscope, however, in 3D more sophisticated imaging systems are required, such as confocal and light sheet microscopy.
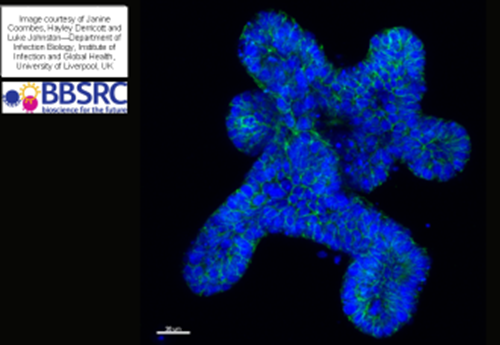
Another novel 3D cell culture strategy is Organoid Culture. Organoids are miniature, simplified versions of an organ produced in vitro in 3D6. They show realistic micro-anatomy and can be derived from one or a few cells from a tissue, embryonic stem cells or induced pluripotent stem cells, which can self-organize, self -renew and differentiate in 3D culture. They have many applications in the field of disease modelling, cell based therapies, drug discovery, cancer biology and in infection studies and there are many examples of their use in the literature; organoids have been developed from the Brain, Lung, Breast, Liver, Pancreas, Intestine, Ovary and Fallopian Tube, Prostate and Stomach.
Organoid culture requires a highly specialised skill set. Media and techniques may not be transferrable between species, for example the same techniques and media may not be appropriate for murine, porcine, bovine or human organoids.
Organoids derived from the gut and airway may carry innate mycoplasma infections and there are challenges associated with cryopreservation protocols and, similar to iPSC cell lines, the standardisation of vial contents may be difficult to achieve. Organoids are dynamic and organo-typic with normal levels of apoptosis and compensated cell death meaning that cell viabilities may be significantly lower than those observed in conventional cell culture.
In summary in order to achieve standardisation and reproducibility in cell culture labs, it is important to have evidence based standard operating procedures and effective training programmes. It is critical to monitor and test cell line authenticity, sterility and mycoplasma status and ensure cell viability at all stages.
As technologies advance, good cell culture guidelines need to evolve to ensure that novel cell culture models are fit for purpose.
References
1.Coecke, S. et al. Guidance on good cell culture practice. a report of the second ECVAM task force on good cell culture practice. Altern. Lab. Anim. ATLA33, 261–287 (2005).
2.Geraghty, R. J. et al. Guidelines for the use of cell lines in biomedical research. Br. J. Cancer111, 1021–1046 (2014).
3.Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science324, 1029–1033 (2009).
4.Takahashi, K. et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell131, 861–872 (2007).
5.Ghaedi, M. et al. Human iPS cell-derived alveolar epithelium repopulates lung extracellular matrix. J. Clin. Invest.123, 4950–4962 (2013).
6.Fatehullah, A., Tan, S. H. & Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol.18, 246–254 (2016).